To celebrate the Year of the Snake, we are delighted to highlight some of the most popular articles published in Inorganic Chemistry Frontiers by corresponding authors based in countries celebrating the Chinese New Year.
We hope you enjoy reading these articles and wish you a happy and prosperous year of the Snake.
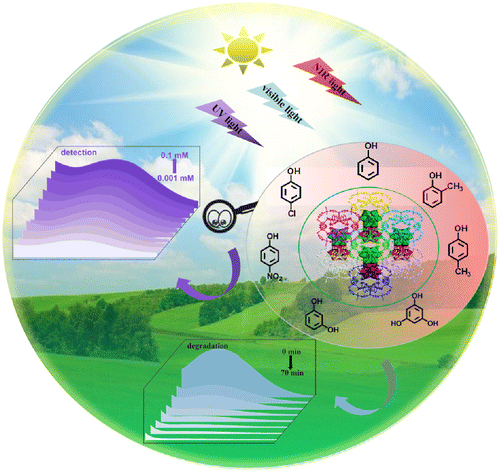 |
Achieving ultra-trace analysis and multi-light driven photodegradation toward phenolic derivatives via a bifunctional catalyst derived from a Cu(I)-complex-modified polyoxometalate
Shuang Li, Bingqian Wang, Guocheng Liu, Xiaohui Li, Chang Sun, Zhong Zhang and Xiuli Wang Inorg. Chem. Front., 2024,11, 1561-1572 |
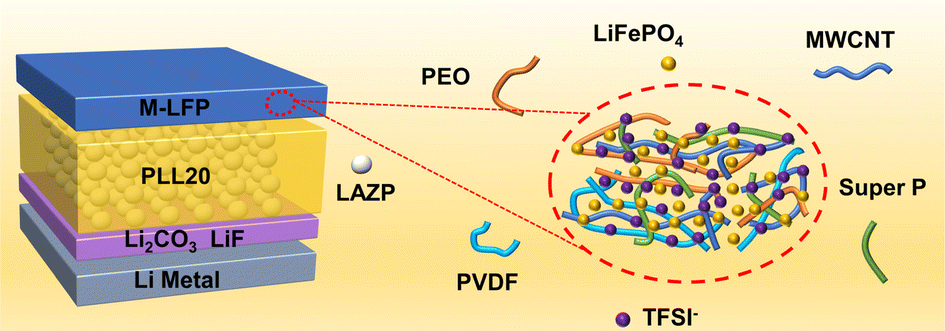 |
PEO/Li1.25Al0.25Zr1.75(PO4)3 composite solid electrolytes for high-rate and ultra-stable all-solid-state lithium metal batteries with impregnated cathode modification
Yongquan Zhang, Hongchang Gao, Jingshun Wang, Qingguo Chi, Tiandong Zhang, Changhai Zhang, Yu Feng, Yue Zhang, Dianxue Cao and Kai Zhu Inorg. Chem. Front., 2024,11, 1289-1300 |
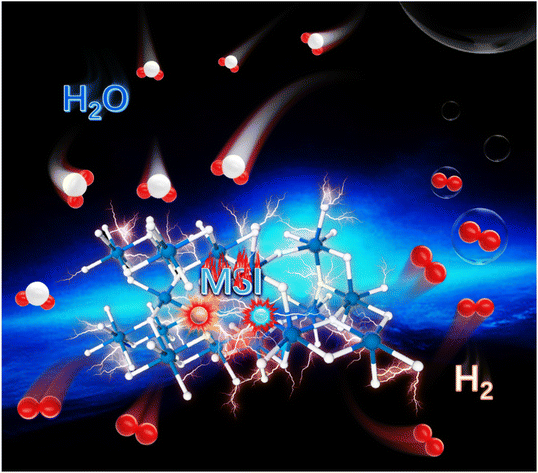 |
Low-content Ru–Pt supported on oxygen vacancy enriched black TiO2 with strong electronic interactions as efficient hydrogen generation electrocatalysts
Yuanzong Shen, Weichen Li, Wenna Wang, Liantao Xin, Weiping Xiao, Guangrui Xu, Dehong Chen, Lei Wang, Fusheng Liu and Zexing Wu Inorg. Chem. Front., 2024,11, 5508-5516 |
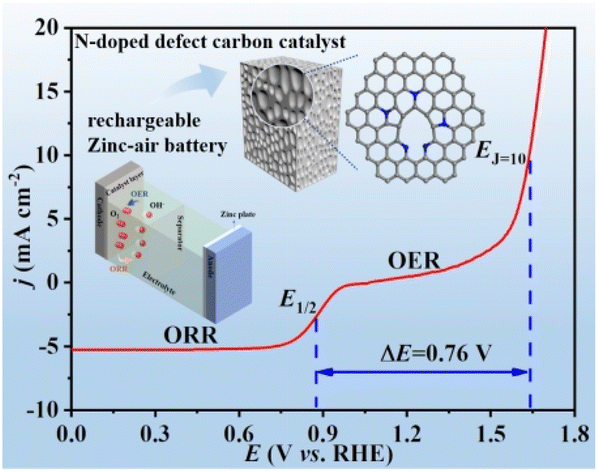 |
Hierarchical mesoporous N-doped carbon as an efficient ORR/OER bifunctional electrocatalyst for rechargeable zinc–air battery
Ping Li, Jinghong Wen, Yang Xiang, Meiqi Li, Yunxiu Zhao, Suna Wang, Jianmin Dou, Yunwu Li, Huiyan Ma and Liqiang Xu Inorg. Chem. Front., 2024,11, 5345-5358 |
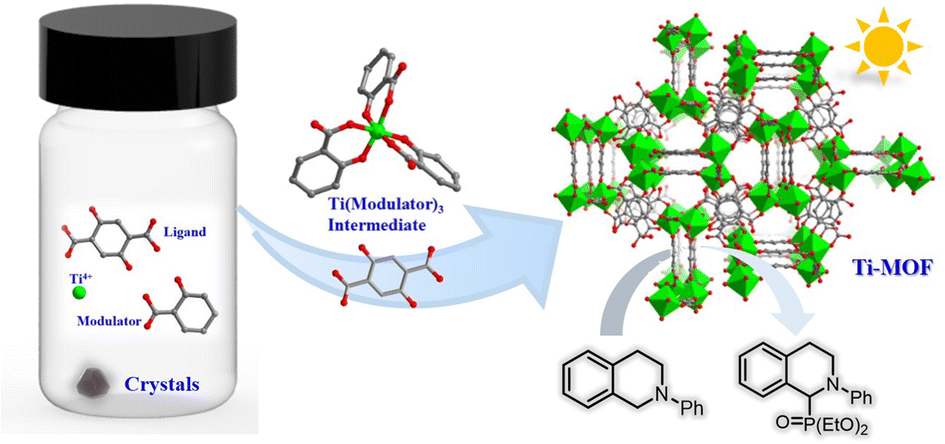 |
A chelating coordination modulation method for the synthesis of Ti-MOF single crystals
Hui-Zi Li, Shangda Li, Fei Wang and Jian Zhang Inorg. Chem. Front., 2024,11, 2876-2883 |
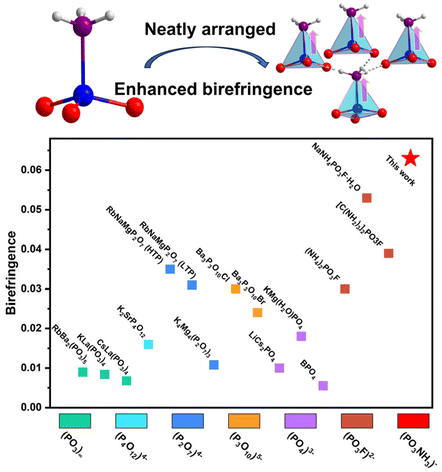 |
Optimized arrangement of non-π-conjugated PO3NH3 units leads to enhanced ultraviolet optical nonlinearity in NaPO3NH3
Lingli Wu, Haotian Tian, Chensheng Lin, Xin Zhao, Huixin Fan, Pengxiang Dong, Shunda Yang, Ning Ye and Min Luo Inorg. Chem. Front., 2024,11, 1145-1152 |
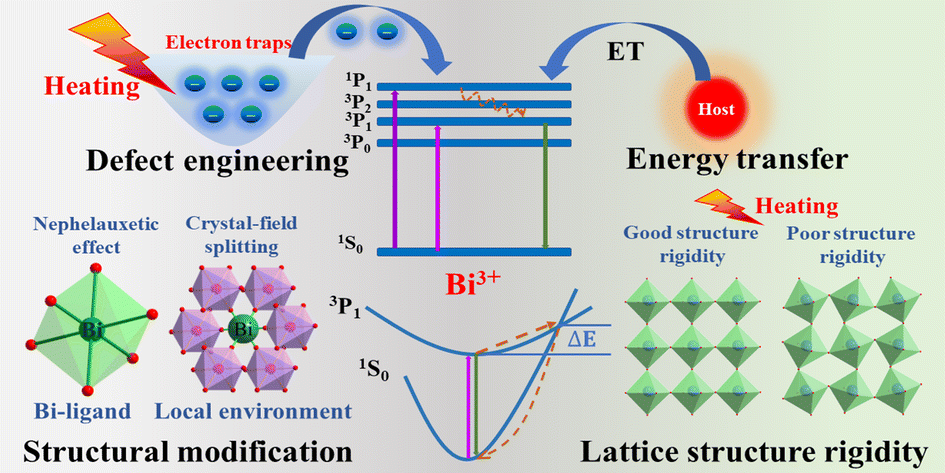 |
Recent progress on modulating luminescence thermal quenching properties of Bi3+-activated phosphors
Xiang Lv, Ran Xiao, Jianxia Liu, Chunwei Yang, Yanmei Xin and Ning Guo Inorg. Chem. Front., 2024,11, 1668-1682 |
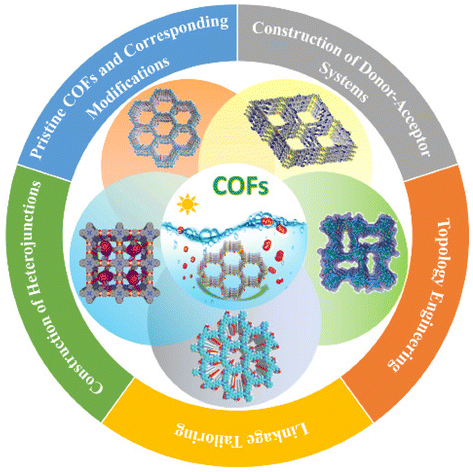 |
Covalent organic framework based photocatalysts for efficient visible-light driven hydrogen peroxide production
Ke-Hui Xie, Guang-Bo Wang, Fei Zhao, Miao-Can Wang, Hao-Yu Zhang, Hao-Ran Ma, Zi-Zheng Chen, Lin Jiang, Yan Geng and Yu-Bin Dong Inorg. Chem. Front., 2024,11, 1322-1338 |
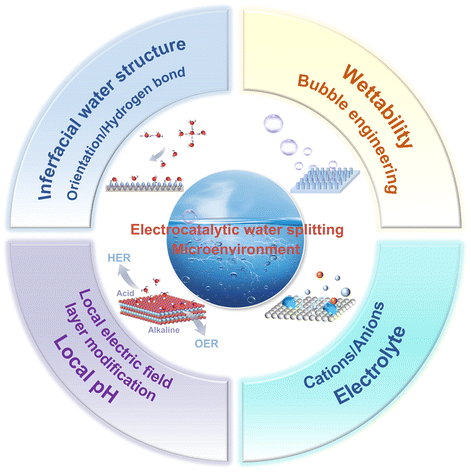 |
Rational design of local microenvironment for electrocatalytic water splitting
Xiang Li, Wangchuan Zhu, Yanqun Zhang, Yueyue Zhao, Danjun Wang, Yanzhong Zhen, Feng Fu and Chunming Yang Inorg. Chem. Front., 2024,11, 4080-4106 |
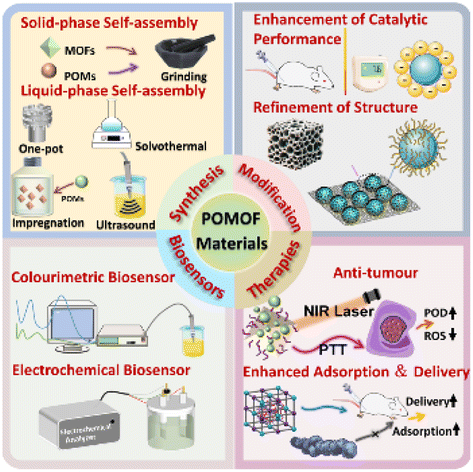 |
Advancing biomedical applications of polyoxometalate-based metal–organic frameworks: from design to therapeutic potential
Lijin Wang, Pengyu Dai, Hongli Ma, Tiedong Sun and Jinsong Peng |








")