Eriko Kamata, Momoko Maeda, Kanako Yanagisawa, Kae Sato*
Department of Chemical and Biological Sciences, Faculty of Science, Japan Women’s University, Bunkyo, Tokyo 112-8681, Japan
*E-mail: satouk@fc.jwu.ac.jp
Why is this useful?
There have been many reports on microfluidic devices for cell culture having upper and lower microchannels separated by a thin PDMS membrane. In these devices, the lower channel often interferes with the microscopic observation of cells cultured in the upper channel. To avoid interference, a microdevice with a detachable lower channel was developed.
What do I need?
Materials:
PDMS (SILPOT 184w/c, Dow Corning Toray)
Hexane
PMMA sheet (56 × 76 × 2 mm)
Glass slides (52 × 76 mm and 26 × 76 mm)
Cover slip (24 × 60 mm)
1 kg weight
PTFE tubing (1 × 2 mm and 0.46 × 0.92 mm)
Tygon tubing (1.59 × 3.18 mm)
Biopsy Punches (1 mm and 2 mm, Kai corporation)
Equipment:
Vacuum desiccator
Oven (65 ˚C and 100˚C)
Spin coater
Plasma generator
Vacuum pump
What do I do?
- PDMS molding
Mix the elastomer and curing agent at a 10:1 mass ratio. De-gas the mixture under vacuum until no bubbles remain (20 min). Pour the degassed PDMS mixture onto a master, which has an upper channel (1 × 1 × 10 mm) or a lower channel (0.5 × 2 × 15 mm) structure, and then place it in an oven at 65˚C for 1 h. Peel off the PDMS replica from the master and adhere it to a glass slide (26 × 76 mm). Place it in an oven at 100˚C for 1 h (Fig.1).
- Preparation of a thin PDMS membrane
Spin coat 600 µL of the PDMS prepolymer (at a 10:1 mass ratio) on a PMMA sheet at 500 rpm for 20 s followed by 2,400 rpm for 600 s. Bake it at 65˚C for 1.5 h.

Fig.1 PDMS sheet with the upper channel, that with the lower channel, PDMS membrane, and tubing.
- Permanent bonding of the PDMS membrane and the sheet with the upper channel
Punch the inlet and outlet holes at both the ends of the upper channel with a 2-mm biopsy punch. Expose both the bonding surfaces of the PDMS membrane and the PDMS sheet with the upper channel (upper sheet) to plasma at 100 W, 35 s (Fig.2a and 2b). Laminate and bake them at 65˚C for 1 h (Fig.2c). Remove the PMMA sheet, and punch a hole to connect the sheet with the lower channel by using a 1-mm biopsy punch from the membrane side.
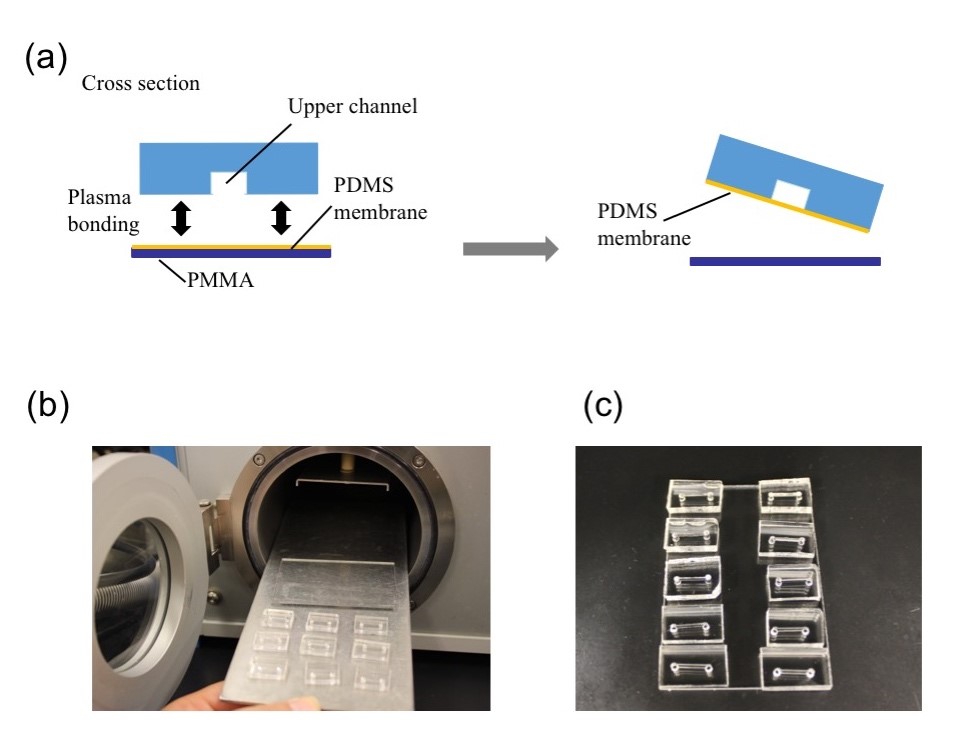
Fig.2 (a) Schematic diagram of bonding the PDMS membrane and the upper sheet. (b) Plasma treatment. (c) Bonded PDMS membrane and sheet.
- Detachable bonding of the PDMS membrane and the PDMS sheet with the lower channel
The PDMS sheet with the lower channel (lower sheet) is bonded to the PDMS membrane by using a PDMS prepolymer diluted with hexane (a dilution ratio of 1:3) as a glue1 (Fig.3a). Spin coat the diluted PDMS prepolymer (600 µL) at 2,000 rpm for 30 s on the surface of a glass slide (52 × 76 mm) to cover the slide with a thin layer of the glue and incubate it for 10 min to dry the solvent (Fig.3b). Place the lower sheet on the coated glass slide (Fig.3c). Apply glue at the four corners of the PDMS membrane bonded with the upper sheet (Fig.3d). Peel off the lower sheet from the glass slide and place the glue-coated surface of the sheet on the PDMS membrane (Fig.3e). After 30 min of incubation, bond the lower sheet to a cover slip by plasma bonding. Place a 1-kg weight and a glass slide on the device and bake it at 100˚C for 1 h (Fig.3f).
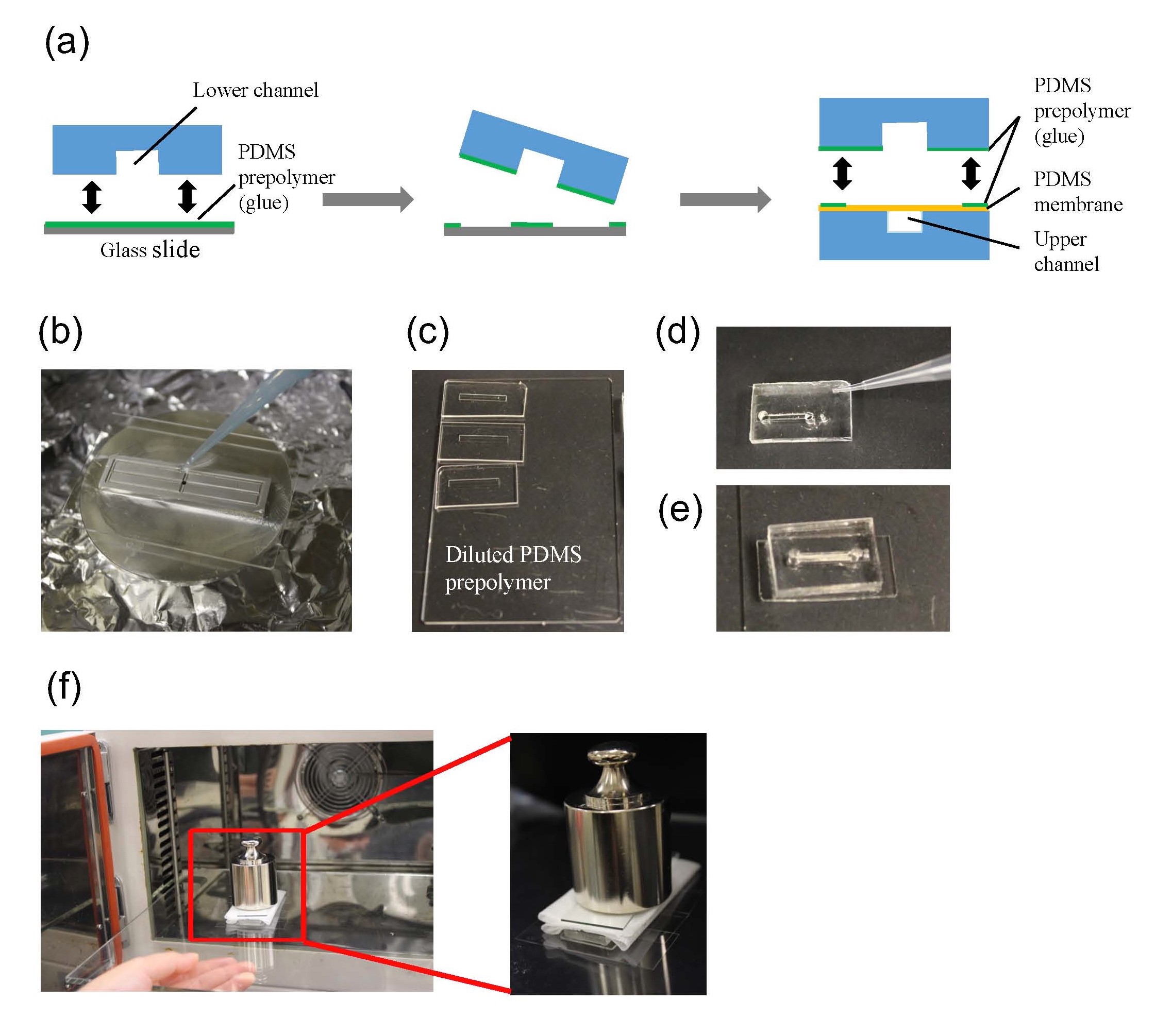
Fig.3 (a) Schematic diagram of bonding of the PDMS membrane and lower sheet. (b) Spin coat the diluted PDMS prepolymer (glue). (c) Lower sheet is placed on the thin film of the diluted PDMS prepolymer. (d) The diluted PDMS prepolymer is applied on the four corners of the PDMS membrane. (e)The glue-coated surface of the lower sheet is placed on the PDMS membrane. (f) A weight and a glass slide are placed on the device to bake it at 100˚C.
- Tubing
Connect polytetrafluoroethylene (PTFE) tubes (1 × 2 × 100 mm) with Tygon tubes (1.59 × 3.18 × 10 mm) to the holes present at both ends of the upper microchannel. Connect a PTFE tube (46 × 0.92 × 150 mm) to the hole of the lower channel. Apply PDMS prepolymer at the root of the tubes, and then bake it at 100˚C for 1 h for firm connection (Fig.4a and b).
- Cell culture
Introduce a cell suspension into the upper microchannel, which is manually precoated with 0.1 mg/mL of fibronectin. Incubate the device at 37˚C with 5% CO2 for 16 h to allow cells to adhere to the bottom of the upper channel (surface of the PDMS membrane).
- Detachment of the lower sheet for cell observation
Remove the lower sheet from the device carefully (Fig.4c and d). Place the rest of the device on a cover slip for observation with an inverted microscope (Fig.4 f and h).

Fig.4 (a) The complete microdevice. (b) Side view of the microdevice. The cell culture channel (upper) is filled with water containing a red food color, while the lower channel is filled with water containing a blue food color. (c) and (d) The lower sheet is peeled off from the microdevice carefully. Phase contrast images of cells (e) before and (f) after detachment of the lower sheet. Fluorescent images of cells stained with CellTracker Red CMTPX (g) before and (h) after detachment.
Conclusion
We developed a microfluidic device with a detachable lower microchannel. It is important that different bonding techniques be used for each side of the PDMS membrane. If the lower channel is filled with air and the device is incubated in a CO2 incubator, dew condensation is often observed in the lower channel when the device is taken out from the incubator. The condensation in the lower channel makes observation difficult (Fig.4e and g). This problem was solved with the detachable device.
Acknowledgment
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP16H04170.
Reference
- Chueh, B.H., Huh, D., Kyrtsos, C.R., Houssin, T., Futai, N., Takayama, S. (2007). Leakage-free bonding of porous membranes into layered microfluidic array systems. Analytical Chemistry 79 (9), 3504–3508.








")