What a year 2023 was! CrystEngComm celebrated 25 volumes, over 1.2 million article downloads, over 575 articles published, including 23 Highlight review articles and 59 Communications, from 47 countries with three themed collections and four Editor’s Collections.
A celebration of 25 volumes of CrystEngComm
The publication of the 25th volume of CrystEngComm in 2023 was another milestone for the journal. From the launch of CrystEngComm in 1999 as one of the first peer reviewed online-only chemistry journals, it has moved forward with increasing submissions, from being a journal with no issues, to monthly, then bimonthly and finally weekly publication, publishing almost 15,000 articles in 25 years, featuring authors from 105 countries across six continents. CrystEngComm truly is an international journal with an international readership, authorship and Editorial Board. To mark this exciting milestone for the journal in 2023 a collection of articles and highlights, representing some of the most highly-cited work across the areas of research published in CrystEngComm over the years was collated. Read the collection here.

Editorial Board
We were delighted to welcome two new members to the Editorial Board in 2023: Professor Bin Zhao, Nankai University, China and Professor Changquan Calvin Sun, University of Minnesota, USA.
 Changquan Calvin Sun is Professor of Pharmaceutics at the University of Minnesota, USA, from which he received his PhD. in Pharmaceutics in 2000. After spending 8 years in the pharmaceutical industry, he joined the Department of Pharmaceutics as an Assistant Professor and was promoted to Full Professor in 2017. Professor Sun’s research focuses on efficient formulation design of high-quality tablet products through the appropriate application of materials science and engineering principles. Two main areas of his current research are 1) crystal and particle engineering for superior pharmaceutical properties; and 2) fundamental understanding of pharmaceutical processes, including powder compaction. He is a Fellow of the American Association for the Advancement of Science (AAAS), a Fellow of the American Association of Pharmaceutical Scientists (AAPS), and a Fellow of the Royal Society of Chemistry (RSC). Professor Sun received the 2019 Ralph Shangraw Memorial Award from the International Pharmaceutical Excipient Council (IPEC) and the 2022 David J. W. Grant Distinguished Scholar Award in Basic Pharmaceutics from the National Institute of Pharmaceutical Technology and Education (NIPTE).
Changquan Calvin Sun is Professor of Pharmaceutics at the University of Minnesota, USA, from which he received his PhD. in Pharmaceutics in 2000. After spending 8 years in the pharmaceutical industry, he joined the Department of Pharmaceutics as an Assistant Professor and was promoted to Full Professor in 2017. Professor Sun’s research focuses on efficient formulation design of high-quality tablet products through the appropriate application of materials science and engineering principles. Two main areas of his current research are 1) crystal and particle engineering for superior pharmaceutical properties; and 2) fundamental understanding of pharmaceutical processes, including powder compaction. He is a Fellow of the American Association for the Advancement of Science (AAAS), a Fellow of the American Association of Pharmaceutical Scientists (AAPS), and a Fellow of the Royal Society of Chemistry (RSC). Professor Sun received the 2019 Ralph Shangraw Memorial Award from the International Pharmaceutical Excipient Council (IPEC) and the 2022 David J. W. Grant Distinguished Scholar Award in Basic Pharmaceutics from the National Institute of Pharmaceutical Technology and Education (NIPTE).
 Bin Zhao is a Distinguished Professor at Nankai University. He received his PhD degree from Nankai University in 2004 and has worked as a Full Professor at the Department of Chemistry since 2009. His current research interests focus on the construction of complicated metal clusters and structure, luminescence and catalysis of cluster-based coordination polymers, as well as their applications in the fields of energy, environment and health, such as the conversion and utilization of CO2, water splitting, luminescent probes etc. The related research won the support of the National Outstanding Youth Science Fund. Bin Zhao has published over 180 research papers and has won various awards including the National Hundred Outstanding Doctoral Dissertation Award (2006), the Chinese Chemical Society Prize for Young Scientists (2006), the Program for New Century Excellent Talents in University (2007) and the Youth Science and Technology Innovation Leader (2017).
Bin Zhao is a Distinguished Professor at Nankai University. He received his PhD degree from Nankai University in 2004 and has worked as a Full Professor at the Department of Chemistry since 2009. His current research interests focus on the construction of complicated metal clusters and structure, luminescence and catalysis of cluster-based coordination polymers, as well as their applications in the fields of energy, environment and health, such as the conversion and utilization of CO2, water splitting, luminescent probes etc. The related research won the support of the National Outstanding Youth Science Fund. Bin Zhao has published over 180 research papers and has won various awards including the National Hundred Outstanding Doctoral Dissertation Award (2006), the Chinese Chemical Society Prize for Young Scientists (2006), the Program for New Century Excellent Talents in University (2007) and the Youth Science and Technology Innovation Leader (2017).
CrystEngComm Outstanding Paper Award
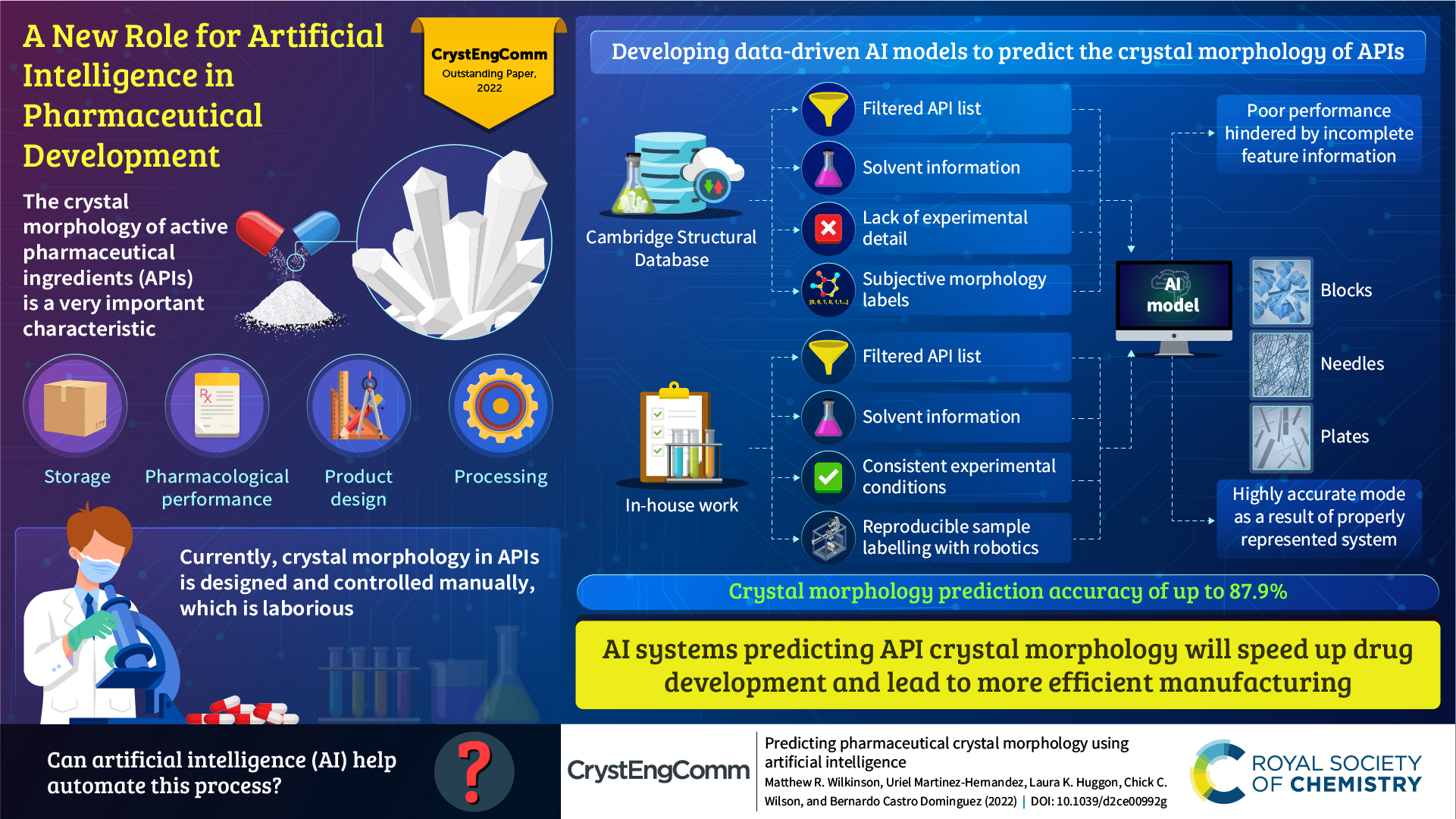
The first Outstanding Paper Award was awarded in 2023 and is a new award aimed at recognising the high-quality work published in CrystEngComm from the previous year, acknowledging the excellence of the paper as a whole and recognising the contributions of all the authors.
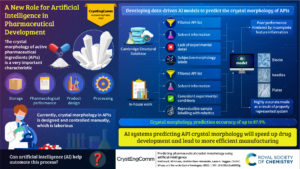
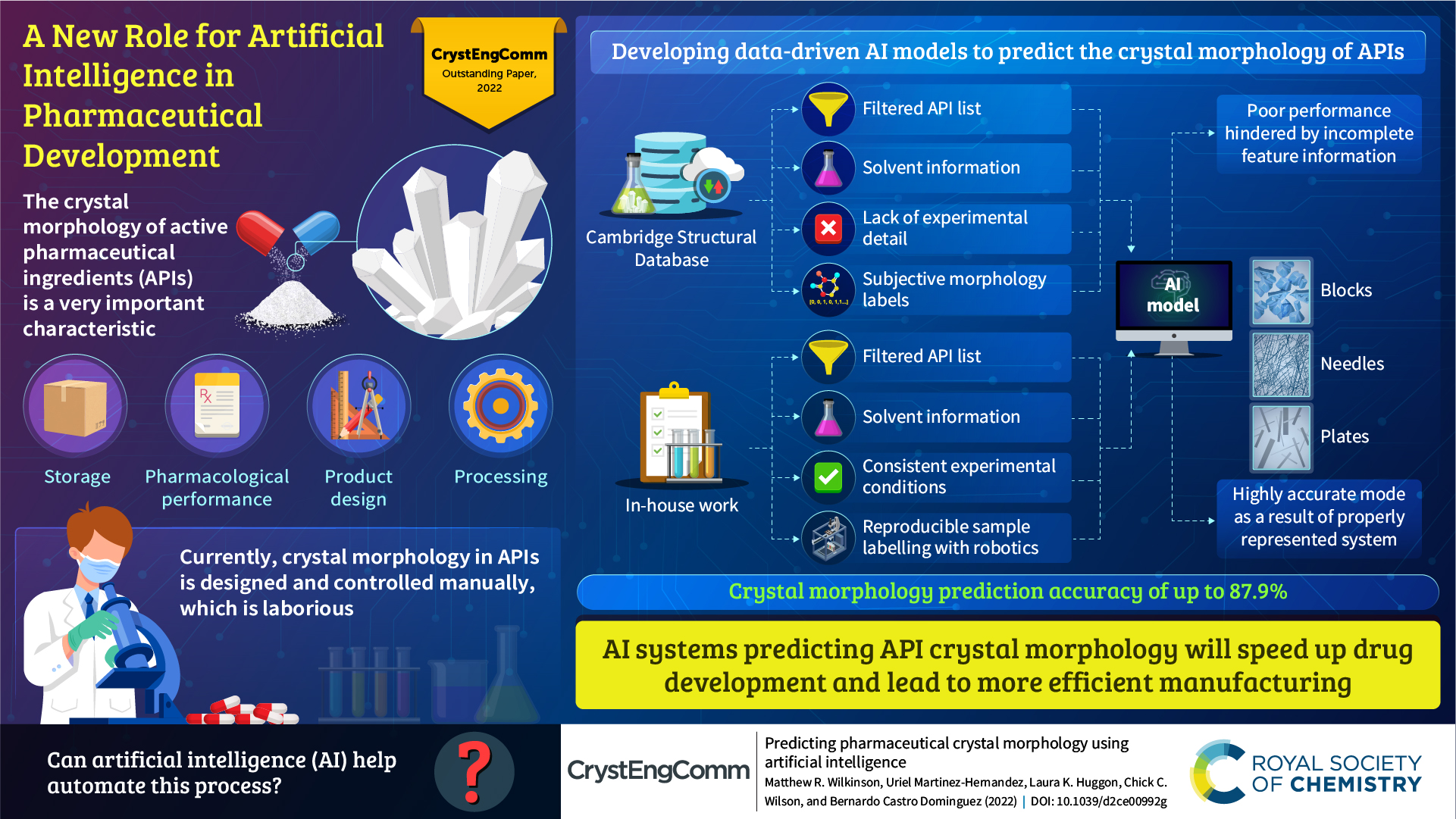
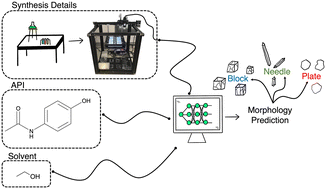
The winners of the CrystEngComm Outstanding Paper Award 2022, as selected by the Editorial Board for their work on Predicting pharmaceutical crystal morphology using Artificial Intelligence, were Matthew R. Wilkinson, Uriel Martinez-Hernandez, Laura K. Huggon, Chick C. Wilson and Bernardo Castro Dominguez.
The authors presented the use of artificial intelligence to predict the morphology of crystallizing active pharmaceutical ingredients, first using publicly available data, and then using their own screening efforts to address the limitations they identified.
Read the article here
CrystEngComm themed collections in 2023
Data Driven Crystal Engineering
This collection, guest edited by Professor Dongfeng Xue and Dr Haitao Zhao, aims to develop the ‘Fourth Paradigm’; revolutionizing crystalline materials R&D methods using advanced data-driven approaches to crystalline materials discovery.
Biomolecular Crystal Engineering
This collection, guest edited by Professor Claudia Pigliacelli and CrystEngComm Editorial Board Chair Professor Pierangelo Metrangolo, features work covering several aspects of crystallization processes involving biomolecules, from the production of single crystals up to their applications as materials in several high-end fields, ranging from catalysis to nanomedicine.
Crystal Engineering in Africa
This collection guest edited by Professor Susan Bourne, Professor Delia Haynes and Professor Patrice Kenfack Tsobnang celebrates the diversity and excellence of research in crystal engineering being carried out across Africa.
Two of our most popular articles published in 2023
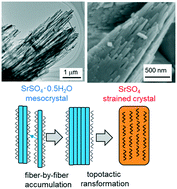
|
|
|
|
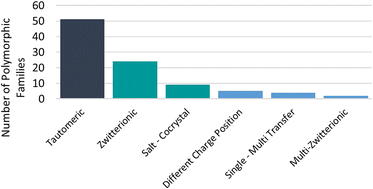
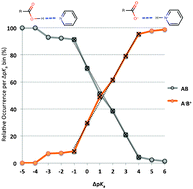
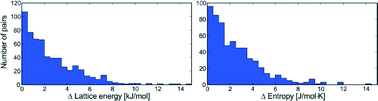
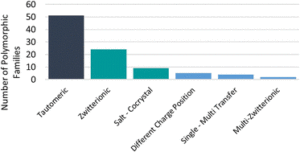
A to Z of polymorphs related by proton transfer
Amy Woods-Ryan, Cheryl L. Doherty and Aurora J. Cruz-Cabeza
CrystEngComm, 2023,25, 2845-2858
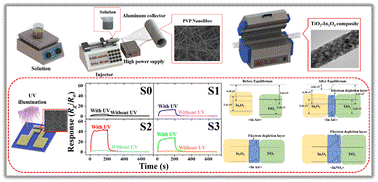
|
 |
| |
|
|
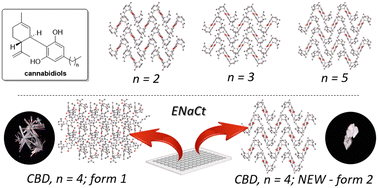
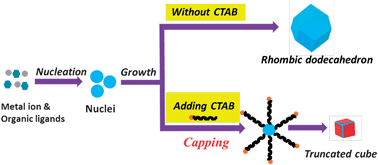
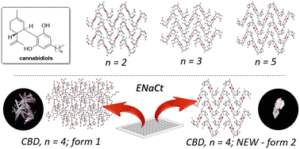
Polymorph prediction through observed structural isomorphism leading to a new crystalline form of cannabidiol
Hannah E. Straker, Lynn McMillan, Lina Mardiana, Glen R. Hebberd, Elle Watson, Paul G. Waddell, Michael R. Probert and Michael J. Hall
CrystEngComm, 2023,25, 2479-2484
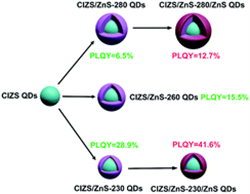
|
 |
CrystEngComm Editor’s Collections, 2023
Curated by Advisory Board members, these collections highlight a number of previously published articles from the journal which the Guest Editor has personally chosen to showcase a specific area of research. The Editor’s Collections published in 2023 are:
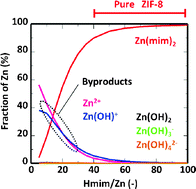
Editor’s Collection: Engineering zeolitic imidazolate framework-8-based materials: This collection of recently published articles focusing on Engineering zeolitic imidazolate framework-8-based materials has been handpicked by CrystEngComm Advisory Board Member, Professor Paolo Falcaro, TU Graz, Austria.
Editor’s Collection: The application of quantum crystallography to solid-state pharmaceuticals: This collection of recently published articles handpicked by CrystEngComm Advisory Board Members, Professor Simon Coles, University of Southampton, UK and Dr Srinivasulu Aitipamula, Institute of Sustainability for Chemicals, Energy and Environment, Singapore, is dedicated to the rapidly-growing field of quantum crystallography and features original research articles from experts in the field, highlighting the latest advancements and future directions of Quantum Crystallography in understanding the structure and properties of pharmaceutical-like materials and organic solids.
Editor’s Collection: Advances in nanocrystal heterojunctions: This collection of recently published articles focusing on nanocrystal heterojunctions has been handpicked by CrystEngComm Advisory Board Member, Professor Georg Garnweitner, Technische Universität Braunschweig, Germany.
Editor’s Collection: Non-classical crystallization processes: This collection of recently published articles focusing on non-classical crystallization processes has been handpicked by CrystEngComm Advisory Board Member, Associate Professor Franca Jones, Curtin University, Australia.
From all the CrystEngComm team, we thank you for your continued interest in and support of the journal.
Comments Off on CrystEngComm: Highlights of 2023
 We have updated our reviewer recommended ‘HOT articles’ for 2024.
We have updated our reviewer recommended ‘HOT articles’ for 2024.







")