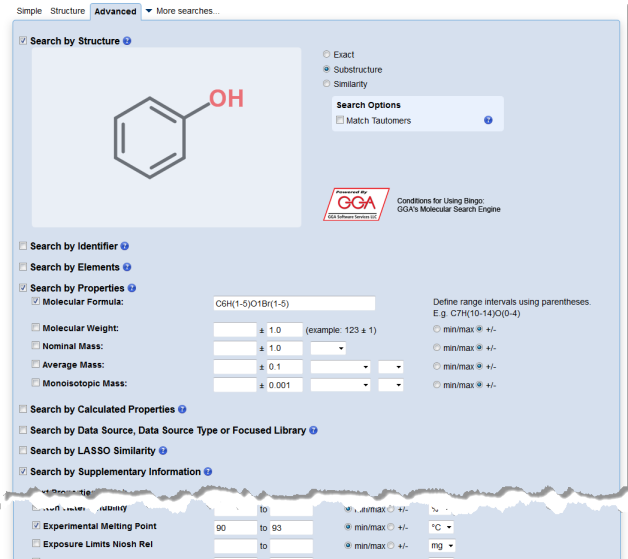
Written by Aileen Day.
This summer there have been a number of students from the University of Southampton doing internships on joint projects between the university and the Royal Society of Chemistry and ChemSpider. Three of these students have been sifting through theses from past members of Richard Whitby’s research group in order to extract the compound, spectra and reaction data in it (and linked lab note books, and archive spectra files) and share these in LabTrove, ChemSpider, and CSSP. The students – Alex Hartke, Yet Wai Lee and Josh Whittam (all 2nd year undergraduates) – are shown below together with the boxes of thesis data, lab notebooks and spectra print outs that they digitised.

Southampton University interns
Between them they digitised 7 theses, by A.Henderson, L. Sayer, D. Owen, D.Macfarlane, F. Giustiniano, G. Saluste, J. Stec, which resulted in 1035 LabTrove pages being published to the Whitby Group’s LabTrove blog.
The theses were a rich source of compound information – including compound structures, names, properties and spectra, all of which were also deposited into ChemSpider resulting in 208 new compound pages, and about 600 spectra.
For this project the students manually deposited the compound information into LabTrove and then deposited the compounds and spectra to ChemSpider. However, we are currently developing a range of ChemSpider jquery widgets which can be integrated into web-based ELNs such as LabTrove which will make it easier to enter compound information from ChemSpider into experiments, and also to publish compound and reaction data from the ELNs to ChemSpider, CSSP and ChemSpider Reactions. This will follow on from the initial proof of concept to retreive ChemSpider information and enter it into LabTrove pages.
With this long-term aim in view, the LabTrove pages that the interns stored the compound and reaction data were structured using LabTrove templates, and this structuring will make it easier for publishing widgets to understand the data and process it the correct way. In this way, the project was partly a test to ensure that the templates were suitable for storing compound data in LabTrove. As well as the ChemSpider compound and associated data template (with corresponding help page, templates were also written to store reaction data in a formatted way, since the theses were primarily focused on the synthesis of compounds. At their simplest, basic reaction data can be stored in LabTrove using the ChemSpider Reactions template (and corresponding help page, and eventually posts written in this format will be easily publishable to ChemSpider Reactions. More detailed reaction data can be stored using the ChemSpider SyntheticPages style reaction template (and corresponding help page. The initial aim was to deposit all of this reaction data into ChemSpider SyntheticPages but it became clear that it was difficult for anyone other than the researcher who conducted the reaction, or their superviser to supply the necessary level of detail for CSSP submissions, and in particular couldn’t easily be reached by retrospectively abstracting theses. As a result, only a handful of reactions were submitted to CSSP, and the majority (over 500) were stored in LabTrove for future submission to ChemSpider Reactions.
If reactions can be published easily from ELNs to ChemSpider Reactions and that is easily queryable by other researchers and their applications when performing new reactions this will be a major step towards the aims of the Dial-a-molecule (an EPSRC Grand Challenge network). An important part of the reaction data which needs to be captured is the stoichiometry table of substances used and produced in a reaction. However, these stoichiometry tables are too complicated to incorporate into a LabTrove template, so the LabTrove reaction templates will be used in conjunction with a new ChemSpider jquery widget which is currently in the process of being integrated with LabTrove (more details to follow on this blog shortly!) which will construct them. The widget performs ChemSpider lookups to retrieve compound information, and will calculate equivalents, thereby saving the researcher time when working out the amounts of reactants needed or yields of products obtained. An example of a reaction post which was initially created using the ChemSpider Reactions template and then supplemented by adding a stoichiometry table to it using the ChemSpider Edit Stoichiometry Table widget is shown here.
If you are a LabTrove user and wish to use the ChemSpider templates, their source is available via their links above, and instructions for using templates in Labtrove are documented here.