Ring-opening polymerizations produce commercial polymeric materials including epoxy resins, but they usually liberate small molecules such as the greenhouse gas, CO2. In the context of climate change, it is urgent to reduce CO2 emissions. Recently, a group of UK researchers led by Prof. Charlotte K. Williams at the University of Oxford developed a step-growth polymerization method that self-consumed CO2. The work has been published in a recent issue of Chemical Communications.
The synthesis involved two catalytic cycles (Figure 1). The first cycle polymerized L-lactide-O-carboxyanhydride into poly(L-lactide acid) (PLLA) via a ring-opening polymerization and released one CO2 molecule per polymer repeat unit. In the second cycle, epoxide molecules (cyclohexeneoxide) combined with the CO2 generated in the first step and grew into poly(cyclohexene carbonate) (PCHC) from the terminal ends of the PLLA chains. A di-zinc-alkoxide compound catalyzed both cycles and coupled the two processes together. The product is PLLA-b-PCHC block copolymers, which are composed of PLLA and PCHC covalently tethered together.
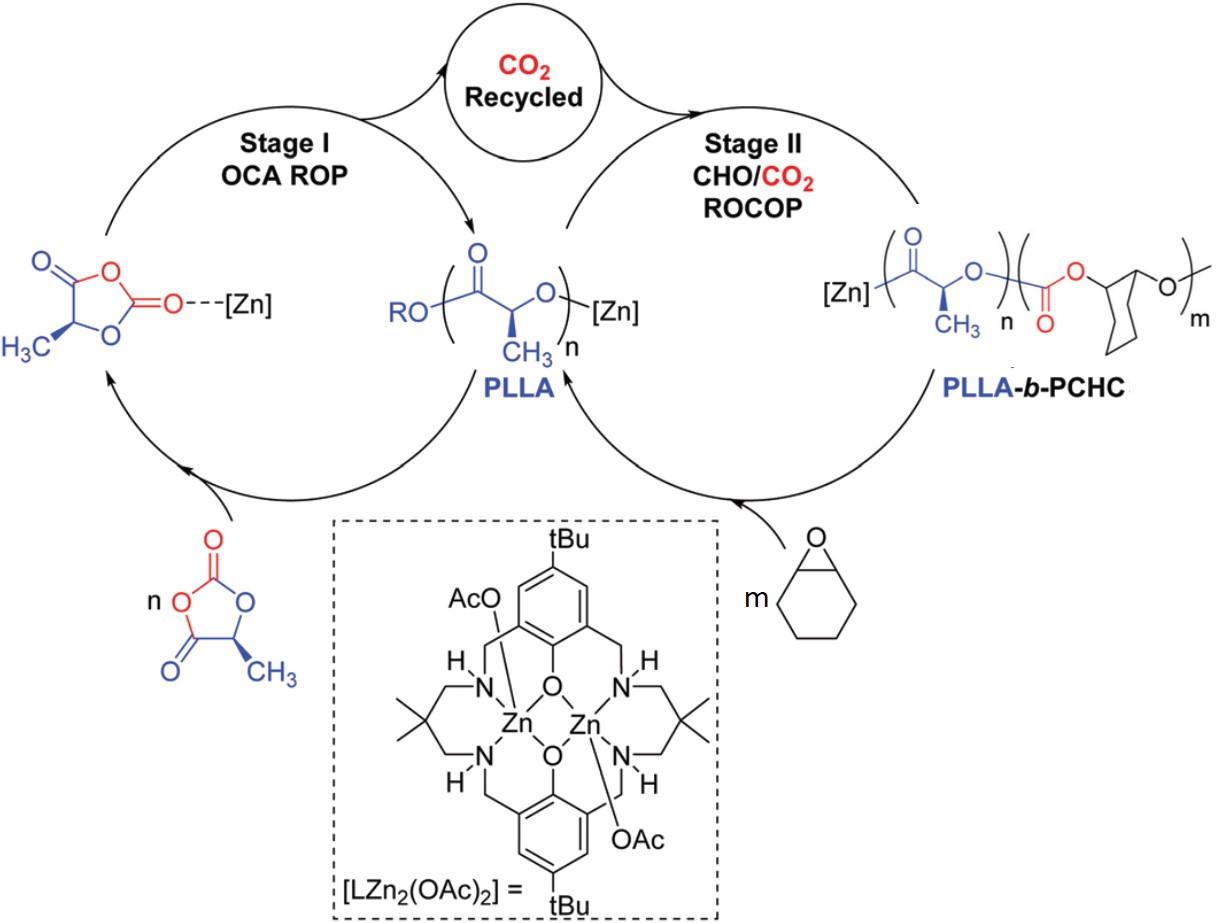
Figure 1. The two catalytic cycles are joined by a zinc-based catalyst, [LZn2(OAc)2]. The CO2 gas produced in the first step serves as a reactant in the second step. OCA: O-carboxyanhydride; ROP: ring-opening polymerization; CHO: cyclohexeneoxide; ROCOP: ring-opening copolymerization.
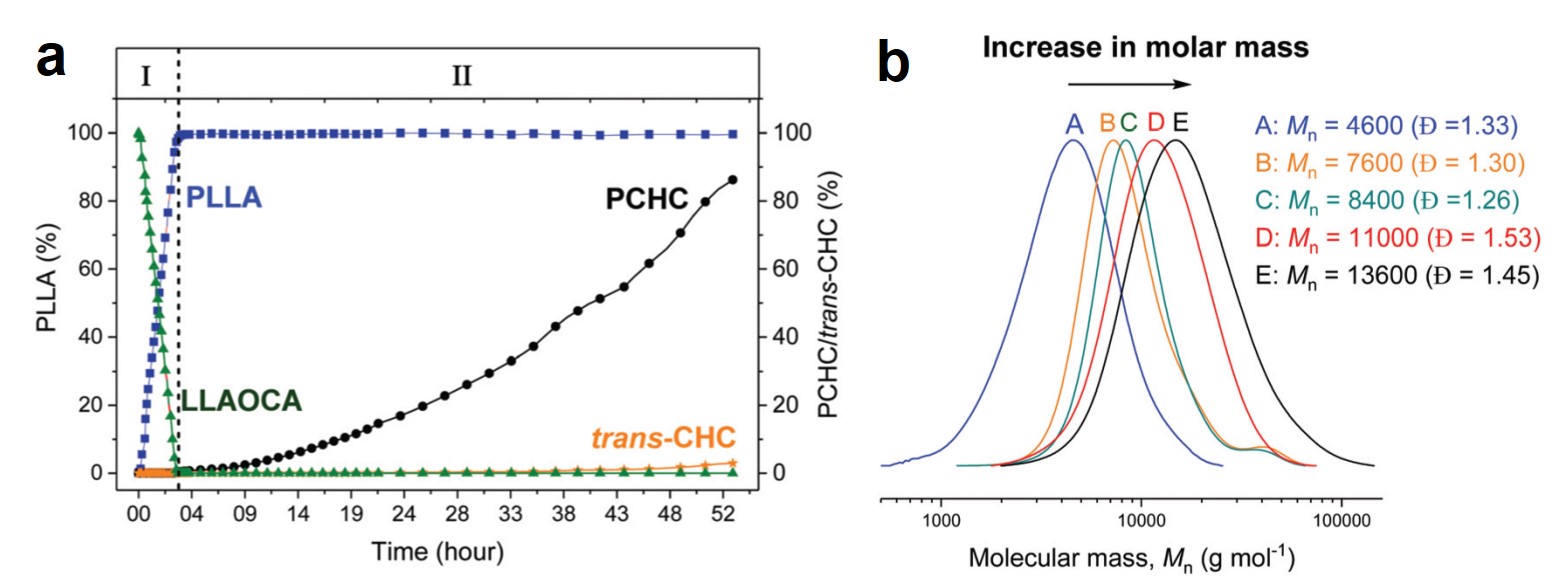
Figure 2. (a) The evolution of the concentrations of PLLA, PCHC, and trans-CHC (the byproduct of the second step) with reaction time. (b) Size-exclusion chromatograms of the products at different reaction times. Mn: number-average molecular weight; Đ: polydispersity.
The authors are investigating the detailed polymerization mechanism, as well as identifying new catalysts to expand the polymerization scheme to other polymers.
To find out more, please read:
Waste Not, Want Not: CO2 (Re)cycling into Block Copolymers
Sumesh K. Raman, Robert Raja, Polly L. Arnold, Matthew G. Davidson, and Charlotte K. Williams
Chem. Commun., 2019, 55, 7315-7318
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from University of California, Santa Cruz in the United States. He is passionate about scientific communication to introduce cutting-edge research to both the general public and scientists with diverse research expertise. He is a blog writer for Chem. Commun. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")