A small consolation when marking undergraduate organic chemistry exams is that occasionally you come across an answer so ridiculous it is almost brilliant. Penned to complete a far-fetched synthesis, the student managed to propose a reaction that is completely without precedent, not only in the course, but in the chemistry literature as a whole. Sadly for the student it is completely wrong, 0 marks.
I imagine that 50 years ago if an undergraduate student had proposed a one-step synthesis of γ-lactams starting with a linear alkylamine, which proceeded by clipping off an N-H bond and a C-H bond, then stitching it together with a carbon monoxide molecule at the junction, they too may have got 0 marks. Yet Matthew Gaunt and researchers in his laboratory at the University of Cambridge have achieved just this via palladium-catalysed C-H activation.
Transition metal-catalysed C-H activation refers to the cleavage of a C-H bond by a transition metal, followed by functionalisation of the metal-bound organic fragment and regeneration of the catalyst. This strategy is counter to the classical approach of organic synthesis: construction of molecular complexity by installing and manipulating reactive functional groups. The object of pre-functionalisation is two-fold: it makes a molecule more reactive (for example, installation of a halide can enable oxidative addition to a transition metal or substitution by a nucleophile) and it directs reactivity to a specific location in the molecule under construction. For C-H activation the challenge is to promote reaction of thermodynamically and kinetically stable C-H bonds, and achieve site-selectivity in a molecule containing many chemically-similar C-H bonds.
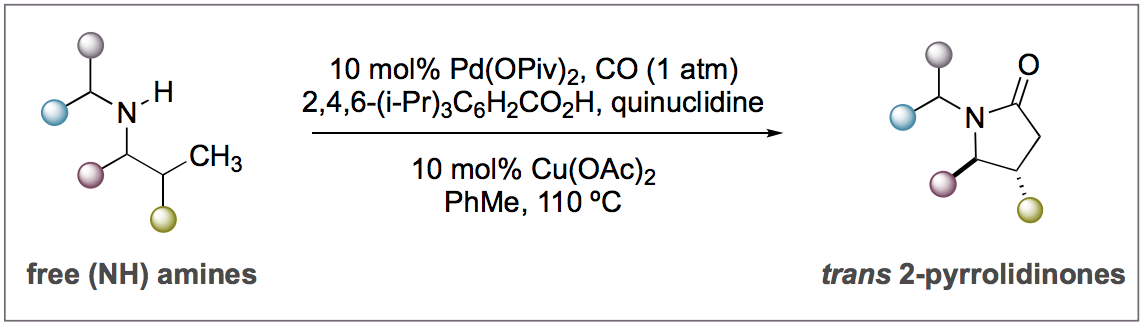
Figure 1: Optimised reaction conditions
The authors found that a catalytic system consisting of palladium pivalate and copper acetate, in combination with acidic and basic additives under a CO/air atmosphere, transformed a variety of secondary amines with primary C-H bonds at the γ-position into 5-membered lactones (Figure 1). Good yields and diastereoselectivities were obtained, and a variety of substituents such as carbocycles, tetrahydropyran, piperadine, fluorocycloalkanes and dioxolanes were well tolerated.
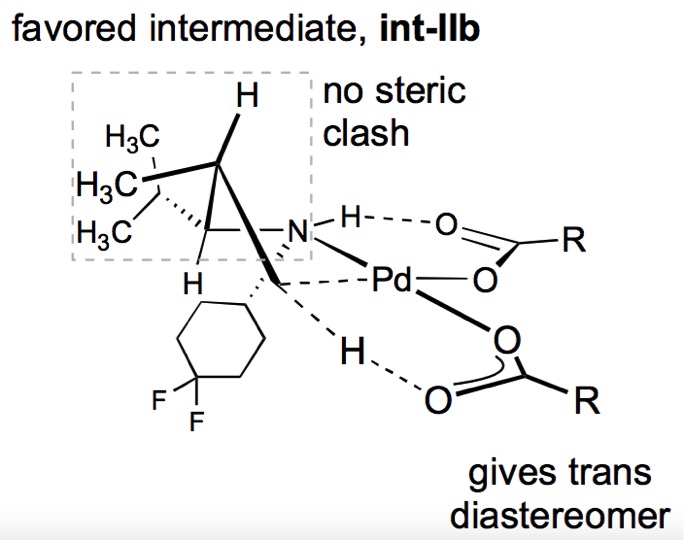
Figure 2: Mechanistic hypothesis showing organisation of the transition state and C-H activation.
The reacting components in the C-H activation step are highly organised in the transition state by coordination of the amine to the palladium centre, and formation of a hydrogen bond between the amine and the carbonyl group of a pivalate ligand bound to palladium (Figure 2). Palladium insertion into the C-H bond (one of the pivalate ligands serves as an intramolecular base) forms a palladacycle with the entropic and enthalpic preference for a 5-membered ring, necessitating abstraction of a proton in the γ-position with respect to the amine directing group.
C-H activation has been referred to as the ‘holy grail’ of catalysis, and the efficiency gains are clear: reduction in reaction steps and use of catalysis minimises energy use, formation of stoichiometric by-products and waste from isolation and purification processes, excess reagents, solvents and additives. This aside, the most exciting thing about the development of C-H activation methods is the promise of discovery: novel reactivity can lead to novel products, inaccessible by other means.
To find out more please read:
Png Zhuang Mao, Jaime R. Cabrera-Pardo, Jorge Piero Cadahia and Matthew J. Gaunt.
Chem. Sci., 2018, Edge Article
DOI: 10.1039/c8sc02855a
 About the author
About the author
Zoë Hearne is a PhD candidate in chemistry at McGill University in Montréal, Canada, under the supervision of Professor Chao-Jun Li. She hails from Canberra, Australia, where she completed her undergraduate degree. Her current research focuses on transition metal catalysis to effect novel transformations, and out of the lab she is an enthusiastic chemistry tutor and science communicator.








")