 Electron paramagnetic resonance (EPR) is powerful technique for studying chemical species containing unpaired electrons. It has far reaching applications in a number of fields as it can be used to elucidate structural, electronic and conformational dynamic features in a given system.
Electron paramagnetic resonance (EPR) is powerful technique for studying chemical species containing unpaired electrons. It has far reaching applications in a number of fields as it can be used to elucidate structural, electronic and conformational dynamic features in a given system. 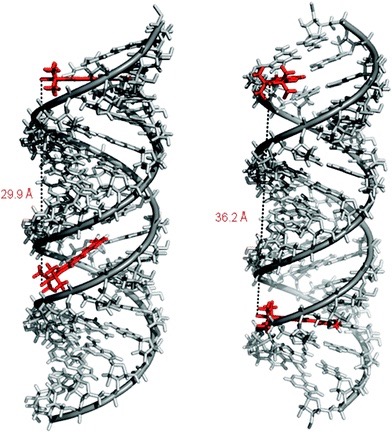
In biological settings, paramagnetic probes have been developed to ‘spin-label’ desired biomolecules using a technique called site-directed spin labelling (SDSL). In combination with other methods, EPR has emerged as an efficient means of studying proteins close to their native physiological states and can be used to glean information regarding the immediate environment of the spin-labeled side-chain as well as measure intra- and intermolecular distances within the protein. A challenge has been in developing rigid spin-labels to improve the accuracy of distance measurements as the most reliable information is attained if the distance between spin-labels is unchanged by conformational flexibility.
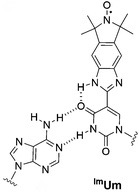
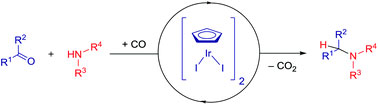
In their most recent OBC publication, Prof. Snorri Sigurdsson of the University of Iceland and Prof. Thomas Prisner of Goethe University describe the development of an enhanced isoindoline-nitroxide derivative of uridine (ImUm), the first example of a conformationally constrained spin label for RNA.
Limited mobility of ImUM is a result of the nitroxide N-O bond lying in the same axis as the bond used to link it to the uridine base. As a result, bond rotation does not drastically alter the position of the nitroxide. Additionally, ImUm was shown to bind specifically and with high affinity to abasic sites in duplex RNAs. Here, rigidity is further enhanced through intramolecular hydrogen bonding between the nitroxide probe and the orphaned uracil base. ImUm is a promising label for EPR studies of RNA, providing highly useful and dynamic structural information unbiased by conformational flexibility.
To find out more see:
A semi-rigid isoindoline-derived nitroxide spin label for RNA
Dnyaneshwar B. Gophane,
DOI: 10.1039/C7OB02870A
Victoria Corless is currently completing her Ph.D. in organic chemistry with Prof. Andrei Yudin at the University of Toronto. Her research is centred on the synthesis of kinetically amphoteric building blocks which offer a versatile platform for the development of chemoselective transformations with particular emphasis on creating novel biologically active molecules.








")
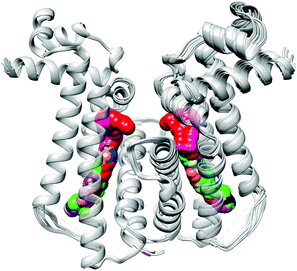
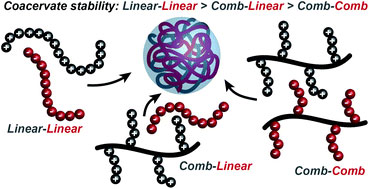 Under favourable conditions, the coalescence of coacervate droplets leads to a separation of a system into two liquid layers: a polymer-rich coacervate phase in equilibrium with a polymer poor supernatant. Coacervation is entropically driven and occurs through an initial electrostatic attraction between oppositely charged molecules followed by the release of counterions and rearrangement of water molecules. However, our understanding of factors that control self-assembly and stability at the molecular level remains limited.
Under favourable conditions, the coalescence of coacervate droplets leads to a separation of a system into two liquid layers: a polymer-rich coacervate phase in equilibrium with a polymer poor supernatant. Coacervation is entropically driven and occurs through an initial electrostatic attraction between oppositely charged molecules followed by the release of counterions and rearrangement of water molecules. However, our understanding of factors that control self-assembly and stability at the molecular level remains limited.