Nature is unrivaled in its ability to produce structurally complex molecules with high biological potency. Natural products have been used medicinally for centuries and have provided a profitable source of potential drug leads. Developing efficient strategies for their total synthesis, as well as the production of analogues, has always been challenging.
In a recent OBC publication, Professor Shuangjun Lin of Shanghai Jiao Tong University has identified a key enzyme in the biosynthesis of the natural product, Streptonigrin, a highly fictionalized aminoquinone isolated from the bacterium Streptomyces flocculus.
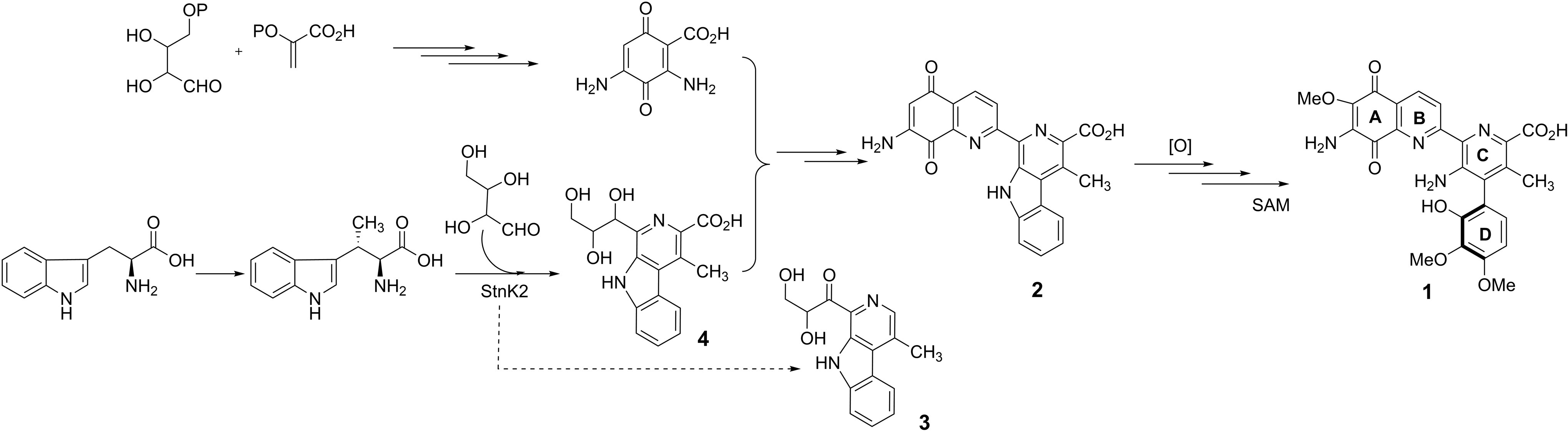
Streptonigrin has a long history and has attracted considerable attention from both the synthetic and biochemical communities due to its challenging molecular framework and potent antimicrobial and broad-spectrum anticancer activities. In the 1970’s, Streptonigrin reached phase-II clinical trials, though ultimately failed due to high levels of toxicity and side effects. Nevertheless, interest in its medicinal properties still remains, and many studies detailing its chemical and biosynthesis have been reported, with the hopes of enabling the production of Streptonigrin-based analogues, which would mitigate the natural product’s cytotoxicity while harnessing is broad biological capabilities.
Prof. Lin reports that previous genetic and biochemical studies successfully identified a key β-carboline (3) intermediate in the biosynthetic pathway of Streptonigrin. The β-carboline moiety is a common structural feature within a large group of natural and synthetic indole alkaloids however, the enzymes catalyzing their formation have not been well characterized or reported. Lin and coworkers have identified a protein, StnK2, which they propose catalyzes a Pictet-Spengler reaction responsible for β-carboline formation from (2S,3S)-β-methyl tryptophan and erythrose (Figure).
The study focussed on the biochemical characterization of StnK2 as a Pictet-Spenglerase, analyzing in detail its stereoselectivity and substrate specificity. StnK2 exhibited exclusive aldehyde specificity, though was flexible towards various tryptophan analogues. Additionally, StnK2 demonstrated high stereoselectivity, only recognizing S-enantiomers and producing the (R)-C-1 of the β-carboline scaffold.
This study has not only contributed to our knowledge of Pictet-Spenglerase enzymes, but has established a new means through which Streptonigrin analogues can be efficiently generated and their medicinal properties explored.
To find out more see:
StnK2 catalysing a Pictet–Spengler reaction involved in the biosynthesis of the antitumor reagent streptonigrin
Xiaozheng Wang, Dekun Kong, Tingting Huang, Zixin Deng and Shuangjun Lin
DOI:10.1039/C8OB02710B
For more papers from the OBC Biosynthesis Themed Collection
Victoria Corless completed her Ph.D. in organic chemistry with Prof. Andrei Yudin at the University of Toronto. Her research centered on the synthesis of kinetically amphoteric building blocks with particular emphasis on creating novel biologically active molecules. She is passionate science and communicating new discoveries to enhance science literacy.








")




{kind=link}