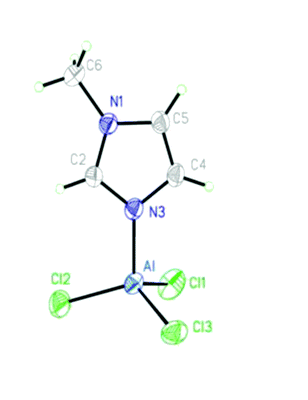
A research team led by Jiang Zhou and Shuquan Liang of Central South University, China, recently identified iron oxyhydroxide (β-FeOOH) as a K-ion battery electrode material. It is reportedly the first iron-oxide-based compound to serve in K-ion batteries. The results have been published in Chemical Communications (DOI: 10.1039/d0cc01009j).
K-ion batteries are a new type of rechargeable battery emerging after Li-ion batteries, the charge-storage functionality of which is associated with the intercalation and de-intercalation of K+. Due to the relative abundance of K+ to Li+, K-ion batteries are poised as a promising alternative to Li-ion batteries.
The researchers made the electrode by a hydrothermal reaction. Specifically, they dispersed Super P® (SP), an electrically conductive additive, into FeCl3 aqueous solutions. The mixture was then heated at 150 °C for 10 h. The resultant powder (FeOOH-SP), comprised of uniformly mixed, crystalline β-FeOOH nanorods and Super P particles (Fig. 1), was directly used as an electrode material.

Figure 1. Transmission electron microscopy images of (a, b) FeOOH-SP and (c) FeOOH. (d) Elemental mappings of C, Fe, and O in FeOOH-SP.
The authors investigated the electrochemical properties of FeOOH-SP in K-ion batteries. At a current density of 100 mA/g, FeOOH-SP exhibited a stable specific capacity of ~200 mAh/g, approximately double and quadruple that of SP and FeOOH alone, respectively (Fig. 2a). The specific capacity of FeOOH-SP was maintained at ~100 mAh/g when the current density increased to 2000 mA/g (Fig. 2b), showing its fast-charging capability. Additionally, the authors observed that the crystalline β-FeOOH nanorods amorphized upon K+ intercalation after being discharged (Fig. 2c). Their crystallinity was only partially restored when being re-charged (Fig. 2d). The loss of crystallinity, however, did not undermine the charge-storage capacity of FeOOH-SP.
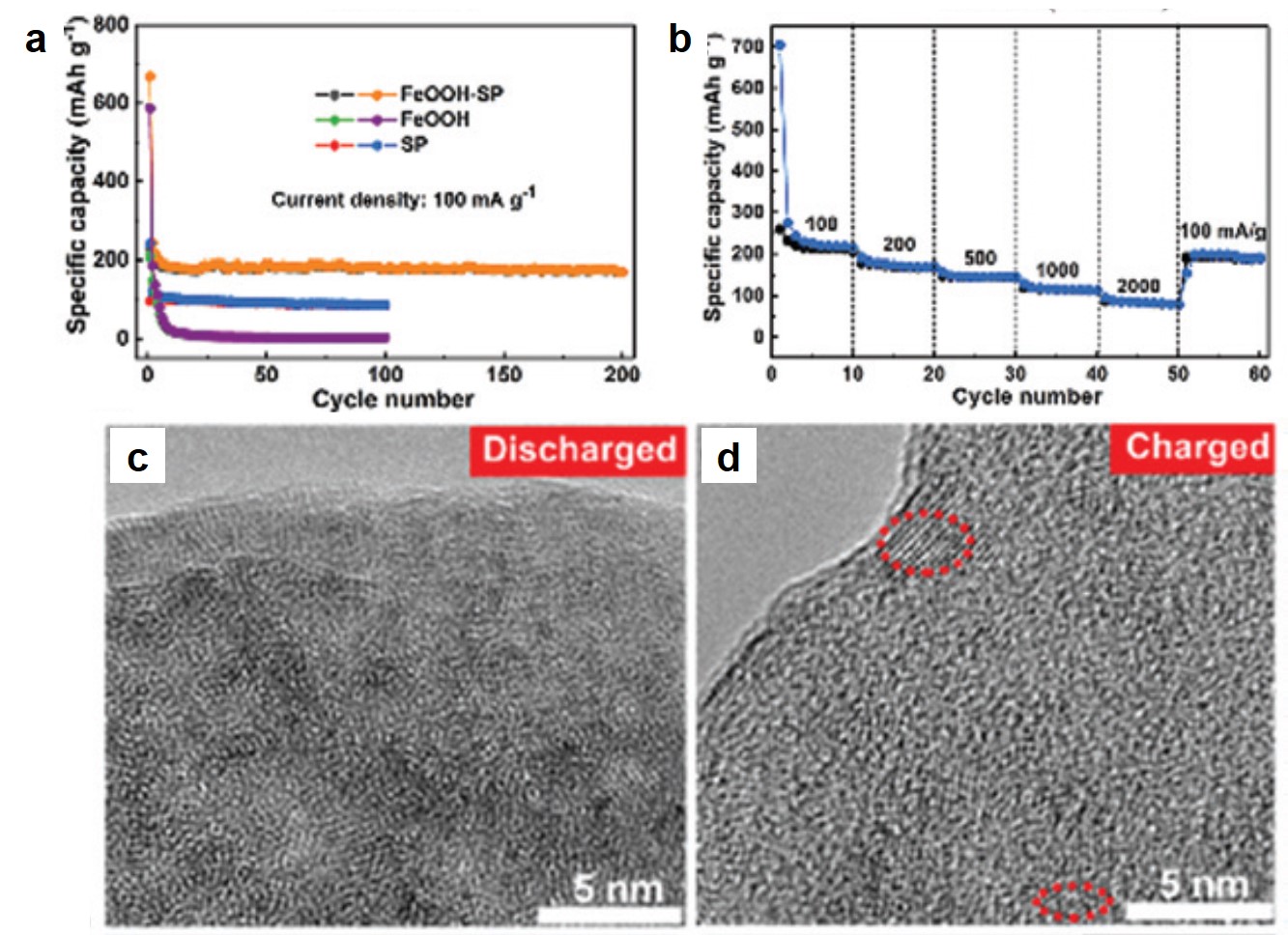
Figure 2. (a) Specific capacities of FeOOH-SP, SP, and FeOOH at different charge-discharge cycles. Current density: 100 mA/g. (b) Rate capability of FeOOH-SP. (c, d) Transmission electron microscopy images of (c) discharged and (d) charged FeOOH-SP. Red dashed boxes highlight crystalline regions. The electrolyte was a mixture of ethylene carbonate and diethyl carbonate containing 1 M potassium bis(fluorosulfonyl)imide.
Considering the low-cost of iron oxyhydroxide, FeOOH-SP could reduce the manufacturing cost of K-ion batteries and increase the affordability of electrochemical charge storage devices.
For expanded understanding, please read:
β-FeOOH: A New Anode for Potassium-Ion Batteries
Xiaodong Shi, Liping Qin, Guofu Xu, Shan Guo, Shuci Ma, Yunxiang Zhao, Jiang Zhou, and Shuquan Liang
Chem. Commun., 2020, 56, 3713-3716.

Tianyu Liu acknowledges Zacary Croft at Virginia Tech, U.S., for his careful proofreading of this post.
About the blogger:
 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at http://liutianyuresearch.weebly.com/.








")