As every chemist knows, carbon likes to play by the rules and is almost always tetrahedral and tetracoordinate. There are exceptions to this geometry though; examples of planar tetra-, penta- and hexacoordinate carbons have been experimentally detected or theoretically predicted. The other group 14 elements also behave similarly to carbon, but again, planar tetracoordinate (pt) or planar hexacoordinate (ph) silicon or germanium atoms have been reported as exceptions to the tetrahedral, tetracoordinate standard.
Planar pentacoordinate (pp) silicon (ppSi) atoms are desirable in 2D materials, and researchers in China and Mexico have now reported the first examples of planar pentacoordinate silicon and germanium atoms (ppX, where X = Si, Ge). The researchers used a ‘π-localisation’ approach in their design, where the formation of multiple bonds between the surrounding ‘ligand’ atoms and the Si or Ge centre led to viable ppX atoms in XMg4Y– (X = Si, Ge; Y = In, Tl) or SiMg3In2 structures (Figure 1).

Figure 1: Structures for the global minima of the two planar pentacoordinate Si/Ge structures
Based on the precedence of B or Al atoms as ligands for stabilising planar hypercoordinate carbon atoms, the researchers first investigated alkaline earth atoms to stabilise the group 14 centres, looking at XM52- structures (X = Si, Ge; M = Mg, Ca, Be). A local energy minimum and a planar, pentacoordinate D5h geometry was only calculated for the triplet-state magnesium species (Figure 2), indicating that analogues of XMg52- were suitable for further investigation.
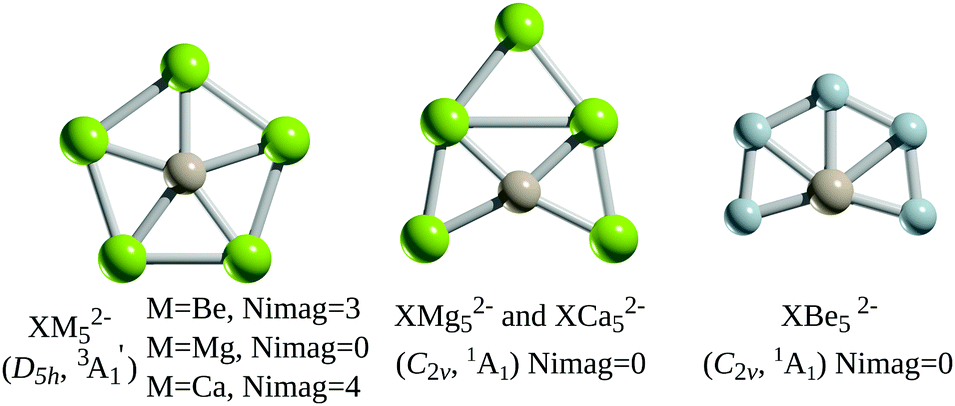
Figure 2: Structures of XM52- (X = Si, Ge; M = Mg, Ca, Be), where Nimag refers to the numbers of imaginary frequencies [3A1 refers to the triplet state, 1A1 refers to the singlet state]
Overall, the researchers have theoretically predicted the first examples of thermodynamically and kinetically stable planar pentacoordinate silicon or germanium atoms. These structures combine both stabilising magnesium and larger group 13 element ‘ligands’, and are suitable candidates for future experimental detection, proving that silicon and germanium can too, break the rules of valency and tetrahedral coordination.
To find out more, please read:
Planar pentacoordinate silicon and germanium atoms
Meng-hui Wang, Xue Dong, Zhong-hua Cui, Mesías Orozco-Ic, Yi-hong Ding, Jorge Barroso* and Gabriel Merino
Chem. Commun., 2020, 56, 13772-13775
About the blogger:
 Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.
Dr. Samantha Apps recently finished her post as a Postdoctoral Research Associate in the Lu Lab at the University of Minnesota, USA, and obtained her PhD in 2019 from Imperial College London, UK. She has spent the last few years, both in her PhD and postdoc, researching synthetic nitrogen fixation and transition metal complexes that can activate and functionalise dinitrogen. Outside of the lab, you’ll likely find her baking at home, where her years of synthetic lab training has sparked a passion in kitchen chemistry too.








")






 Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at
Tianyu Liu obtained his Ph.D. (2017) in Chemistry from the University of California, Santa Cruz, in the United States. He is passionate about the communication of scientific endeavors to both the general public and other scientists with diverse research expertise to introduce cutting-edge research to broad audiences. He is a blog writer for Chem. Comm. and Chem. Sci. More information about him can be found at