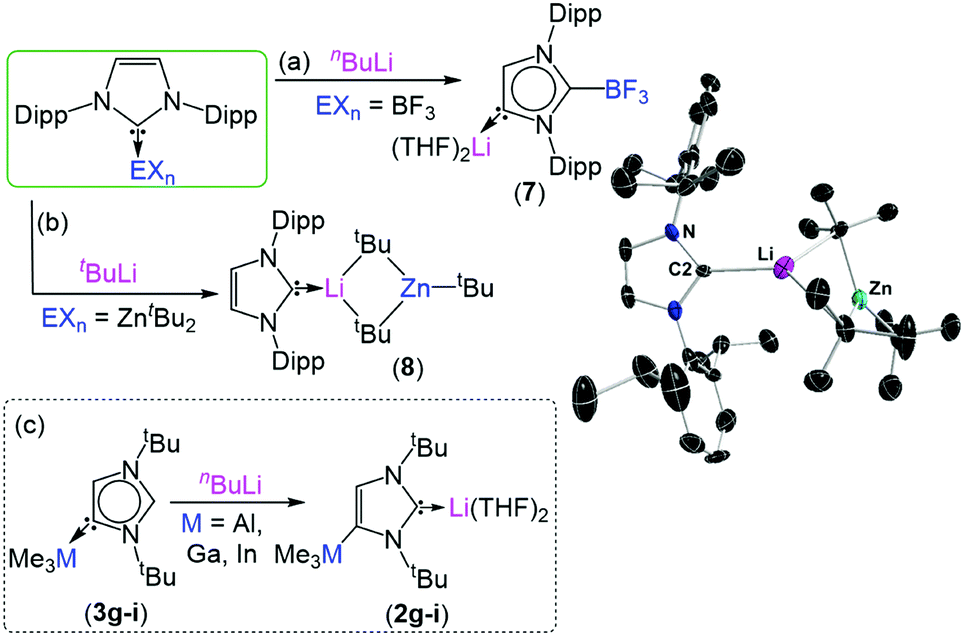
A MOC, I learned this week, is a metal-organic cage. I was familiar with MOMs, MOFs and MOBs, but MOCs were a new one. A MOM (metal-organic material) is a coordination-driven assembly constructed from metal nodes linked by organic ligands. MOMs encompass both MOFs (metal-organic frameworks) and MOCs (metal-organic cages). A MOF is an extended network with the potential for inner porosity, and a MOC is a discrete metal-ligand cluster. And that’s just about as far down the nomenclature rabbit hole I’m willing to go. If you’re keeping up you’ll realise that I forgot one! A MOB is a crowd of graduate students competing for free coffee at the public seminar.
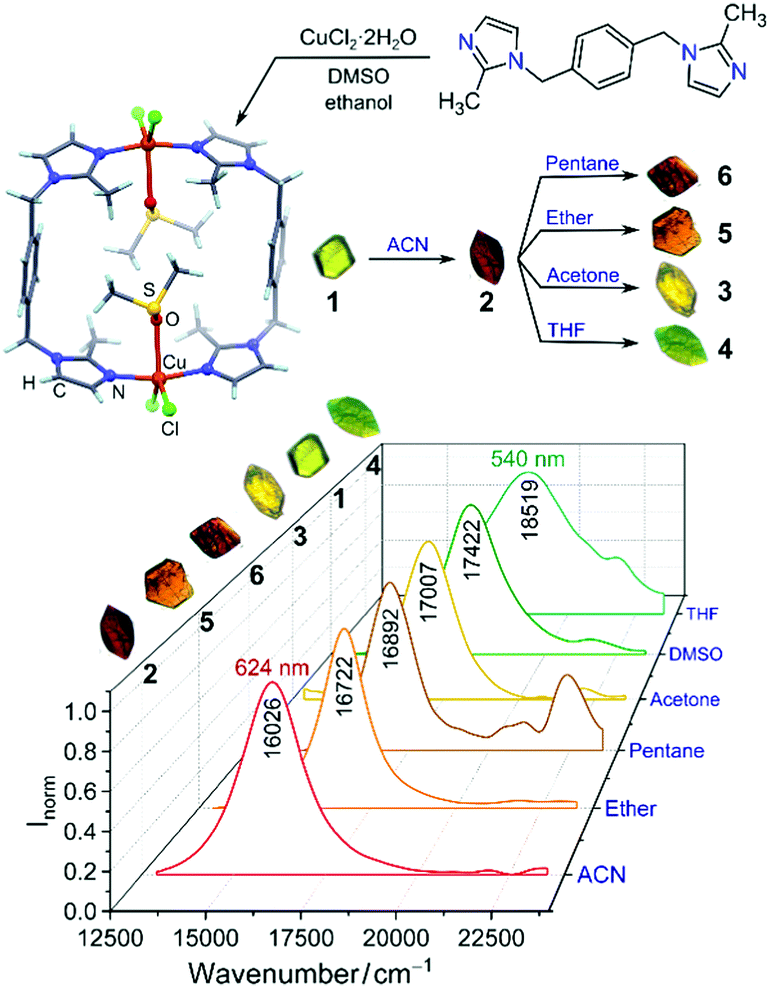
Dong and co-workers at Shandong Normal University designed and prepared a MOM catalyst constructed from copper(II) nodes and a tripodal ligand consisting of a phenylic wheel functionalised with diketones. In chloroform these two components arrange into discrete MOC assemblies containing two tripodal ligands and three copper ions. The copper ions in the cluster are each coordinated to two diketone moieties (in a acetylacetonate-like fashion) in a quasi-square planar arrangement.
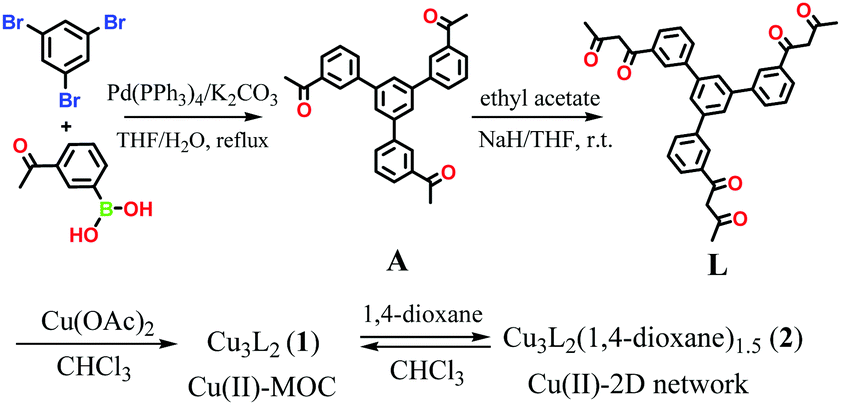
Synthesis of the tripodal ligand functionalised with diketone coordinating moieties.
An interesting property of the material is that it can switch between the MOC form, soluble in halogenated solvents, and an insoluble MOF that assembles upon addition of 1,4-dioxane. 1,4-Dioxane is both an anti-solvent and a ligand; coordination between copper and 1,4-dioxane binds the discrete MOC cages to each other, arranging them into the extended MOF structure. This behaviour can be exploited to prepare a practical catalyst that combines the benefits of both homogeneous and heterogeneous catalysis, namely that homogeneous catalysts are generally more efficient, selective and easier to study, but heterogeneous catalysis are easier to recover and recycle. What better way to solve this problem than with a catalyst that is homogeneous during the reaction conditions, but heterogeneous when it comes to product separation?
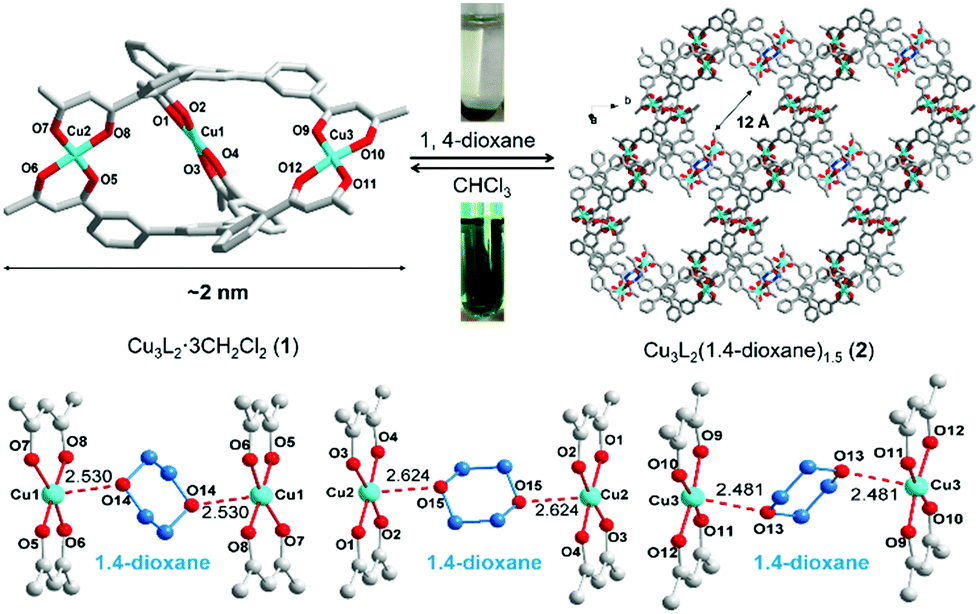
Reversible MOC(top left)-MOF(top right) transition mediated by the addition of 1,4-dioxane. Coordination bonds between 1,4-dioxane shown (bottom image).
The authors used the A3 coupling reaction to demonstrate this concept in a catalytic reaction. The A3 reaction is a transition metal-catalysed, multi-component coupling reaction between aldehydes, alkynes and amines. The products are propargylamines, practical synthetic intermediates for the synthesis of nitrogen heterocycles. The A3 reaction has been extensively studied and can be effected by a wide range of transition metal catalysts. Its versatility makes it a popular choice as a model catalytic reaction to demonstrate innovative ideas in catalytic design – as the authors have done here.
Coordination-driven assemblies have a unique potential for the synthesis of differentially soluble materials, used by the authors for novel catalytic design. Whether this particular metal-ligand combination can be applied to other copper catalysed reactions remains to be seen, nevertheless the principle offers an innovative approach that augments the range of methods striving to bridge the gap between homogeneous and heterogeneous catalysis.
To find out more please read:
Gong-Jun Chen, Chao-Qun Chen, Xue-Tian Li, Hui-Chao Ma and Yu-Bin Dong.
Chem. Commun., 2018, 54, 11550-11553
DOI: 10.1039/c8cc07208f
 About the author
About the author
Zoë Hearne is a PhD candidate in chemistry at McGill University in Montréal, Canada, under the supervision of Professor Chao-Jun Li. She hails from Canberra, Australia, where she completed her undergraduate degree. Her current research focuses on transition metal catalysis to effect novel transformations, and out of the lab she is an enthusiastic chemistry tutor and science communicator.








")
![Synthesis of oxazolines from ketones and isocyanaoacetate esters via a formal [3+2] cycloaddition reaction catalysed by a multicatalytic system of silver and a dihydroquinine squaramide organocatalyst](https://blogs.rsc.org/cc/files/2018/04/Screenshot-2018-04-03-16.24.41.png)